INTRODUCTION
Extrapulmonary small cell carcinomas are uncommon malignant neoplasms with a reported incidence in the United States of 0.1% to 0.4%. They account for 2.5 to 5.0% of all small cell carcinomas. Extrapulmonary small cell carcinomas have been found in nearly every organ system1). They are aggressive neoplasms, which are often disseminated at presentation. Morphologically, immunohistochemically, and ultrastructurally, they are indistinguishable from small cell lung cancer2). The primary locations involved are the salivary glands, thyroid, larynx, trachea, thymus, pleura, esophagus, stomach, intestines, rectum, pancreas, gall bladder, cervix, uterus, breast, prostate, urinary bladder, skin, head and neck3). Moreover, primary small cell carcinoma of the liver is a very rare disease, which requires a careful systemic evaluation and a immunohistochemical examination2). The treatment for unresectable small cell carcinoma of the liver is usually cisplatin or carboplatin-based combination chemotherapy, but survival rates and efficacy are uncertain2).
We report a case of small cell carcinoma of the liver with partial response to systemic chemotherapy.
CASE REPORT
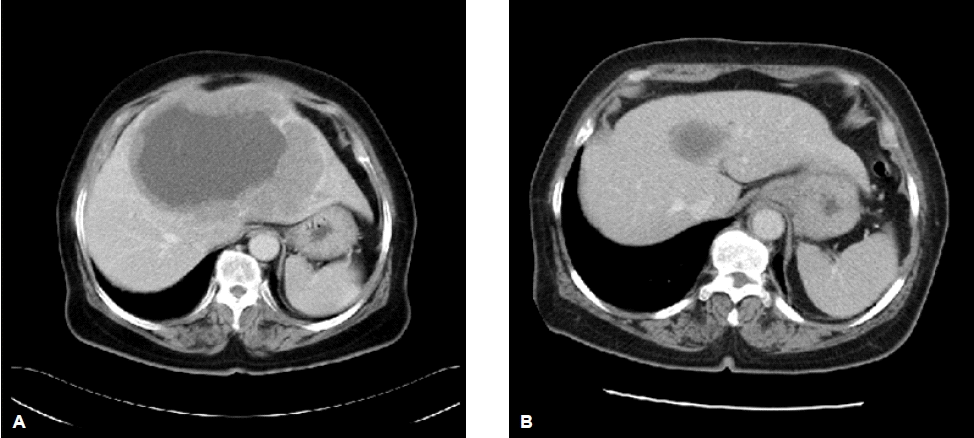
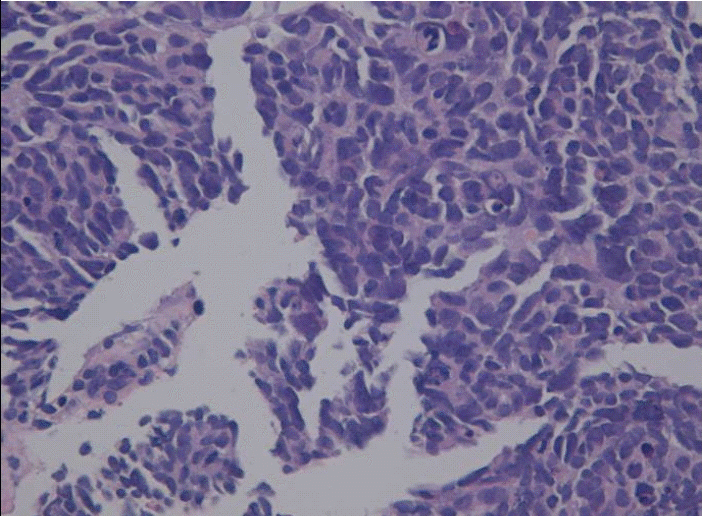
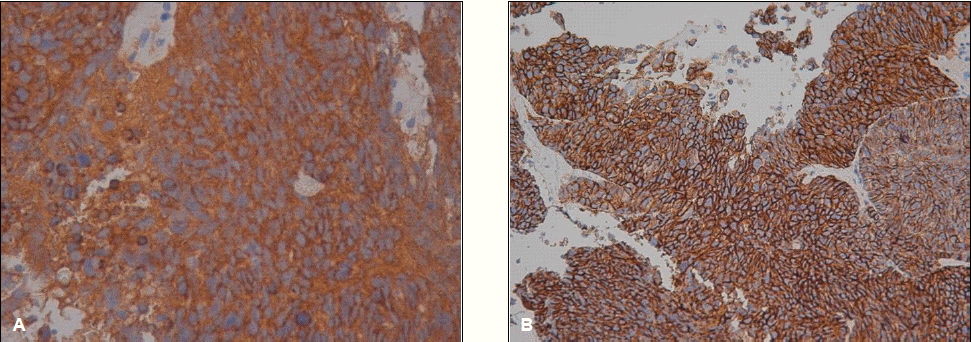
A 65-year-old woman was referred to our hospital to evaluate an abdominal mass. One month before admission, she complained of occasional heartburn. One week before admission, she had no improvement in her symptoms. Therefore, we conducted abdominal sonography and a esophagogastroduodenoscopy, and a 11.5×15 cm-sized liver mass was found. On physical examination, a large mass was palpated in the right upper quadrant without nodularity, while other physical findings were normal. Initial laboratory testing showed that a CBC and the biochemistry levels were within normal limits. Serum levels of alpha-fetoprotein were also within normal limits. Hepatitis B surface antigen, hepatitis B surface antibody, and hepatitis C virus antibody were negative. A computed tomography (CT) scan of the abdomen showed a 15 cm hypervascular mass with central necrosis, internal hemorrhage, and portal vein thrombosis but no underlying liver cirrhosis feature (Fig. 1A, 1B). We performed a needle biopsy of the liver because the CT findings and lab data were not consistent with hepatocellular carcinoma. The histologic diagnosis was small cell carcinoma, which are round to spindled-shaped small cells with dense nuclei, inconspicuous nucleoi, and sparse cytoplasm. A high mitotic index, growth in sheets, a trabecular growth pattern, and apoptosis were shown (Fig. 2). The immunochemical staining was positive for synaptophysin (Fig. 3A) and CD 53 (Fig. 3B) but negative for thyroid transcription factor-1 (TTF-1). Chromogranin and CK7 were focally stained. We performed a systemic evaluation to identify the primary tumor origin.
We could not find any other abnormalities, so she was diagnosed with primary small cell carcinoma of the liver. Systemic chemotherapy was administered consisting of 100 mg/m2 etoposide for 3 days and 75 mg/m2 cisplatin for 1 day. Following five cycles of chemotherapy, the liver mass had decreased from 5 to 3 cm (Fig. 1A, 1B) and the patient was stable during the 12 months. However, the mass progressed to 7 cm, so she was treated with 125 cm/m2 genexol and carboplatin AUC, 5 per day as a second line regimen. After 5 cycles of second line chemotherapy, the 7 cm-sized liver mass had decreased to 3 cm. She made regular follow-ups to the outpatient department for 16 months.
DISCUSSION
Even today, most gastroenteropancreatic neuroendocrine tumors are referred to as carcinoids. The term carcinoid was in-troduced by Oberndorfer in 1907 to reflect carcinoma-like ileal tumors with very low grade malignancy4). In 1963, Williams and Sandler categorized carcinoids based on embryogenetic aspects into foregut (lung, stomach, duodenum, upper jejunum, and pancreas), midgut (lower jejunum, ileum, appendix, and cecum), and hindgut (colon and rectum) carcinoids. This classification was the first to emphasize clinicopathological differences between the tumor groups composing the gastroenteropancreatic neuroendocrine tumors (GEP-NETs) but was never generally accepted in routine diagnostic practice because it was too imprecise to distinguish between the different biologically relevant GEP-NET entities5).
In the first WHO classification of endocrine tumors, published in 1980, the term carcinoid was applied to most neuroendocrine tumors, but this often led to misunderstandings between pathologists and clinicians. One of the main reasons for these misunderstandings was that the pathologists applied the term carcinoid to all tumors with neuroendocrine features, whereas the clinicians understood the term carcinoid to mean a serotonin-producing tumor with carcinoid syndrome5).
For these reasons, the neutral and inclusive terms neuroendocrine tumor and neuroendocrine carcinoma were chosen for the WHO classification of 2000. This WHO classification divided the three entities based on histological features: first, a well-differentiated neuroendocrine tumor, which shows benign behavior or uncertain malignant potential; second, a well-differentiated neuroendocrine carcinoma, which has low-grade malignancy; third, a poorly differentiated neuroendocrine carcinoma of high-grade malignancy. Usually, this third group of tumors are small cell carcinomas5).
Small cell carcinoma can involve the lung, salivary glands, thyroid, larynx, trachea, thymus, pleura, esophagus, stomach, intestines, rectum, pancreas, gall bladder, cervix, uterus, breast, prostate, urinary bladder, skin, head, and neck 3), but a primary neuroendocrine tumor of the liver is very rare. Since primary small cell carcinoma of the liver was first described by Hsueh6) in 1983, only 55 cases have been reported in the English literature. Several theories have been postulated as to the cells of origin, including: (1) a pleuripotential cell or transformation of a liver stem cell, (2) pancreatic rest tissue occurring in the liver, and (3) intrahepatic biliary neuroendocrine stem cells. The favored explanation is differentiation and proliferation from biliary epithelium, because bile ducts contain neuroendocrine argentaffin cells4). Primary neuroendocrine tumors of the liver typically show a solitary mass. However, there are reports of multicentricity of this tumor, mimicking liver metastases4).
The histologic features of small cell carcinoma by light microscopy include a trabecular, pseudoglandular, or insular growth pattern. The cells are characterized by fairly uniform nuclei and abundant granular or faintly staining (clear) cytoplasm. They also contain small clear vesicles, corresponding to the synaptic vesicles of neurons1).
Secretory granules, cytosolic proteins, and small secretory vesicles have been targeted as neuroendocrine differentiation markers. The secretory granule-associated markers include chromogranins A, B, and C (secretogranin II), HISL-19, and CD57(Leu 7). Synaptophysin, synapsin, synaptotagmin, synaptic vesicle protein 2, and synaptobrevin are among the small vesicle-associated markers7). Measuring levels of CD56, CD99, and PHE5 may also be helpful. Typically, a cocktail of markers targeting a secretory granule, a cytosolic protein, and a secretory vesicle is used (i.e., chromogranin A, NSE, and synaptophysin). These markers are helpful to support the hematoxylin and eosin diagnosis of neuroendocrine tumors4).
Surgical excision of the primary neuroendocrine tumor remains the mainstay of therapy for a solitary resectable hepatic mass. For unresectable or recurrent tumors, therapy includes surgical resection, palliative transcatheter arterial chemoembolization, and systemic chemotherapy8).
When the 65-year-old woman visited the local clinic because of epigastric soreness, a 15 cm sized huge liver mass was found during abdominal sonography. The abdominal CT showed a 15cm sized hypervascular hepatic mass containing central necrosis and internal hemorrhage with obliteration of the middle and left hepatic vein. However, her liver function tests, hepatitis viral markers, and ąlpha-fetoprotein levels were within normal limits. A needle biopsy was conducted to disclose the true characteristics of the mass. The histologic findings were no fibrosis in the liver parenchyma or regeneration nodules but round to spindled-shaped small cells with dense nuclei tumor cells showed severe dysplasia with an increased N/C ratio and spare cytoplasm. The mass was positive for synaptophysin and CD 56 immunochemical staining but negative for thyroid transcription factor-1 (TTF-1). Chromogranin and CK7 were focally stained. We did not detect primary lesions in the lungs, stomach, colon, or pancreas with radiologic and endoscopic evaluations. We could not surgically excise the primary neuroendocrine tumor due to the 15 cm sized huge mass and thrombosis in the portal vein.
In general, treatment for an unresectable tumor includes systemic chemotherapy, which has similar response rates to small cell lung carcinoma and reported long-term survivors in other extrapulmonary small cell carcinomas9). There have been no randomized trials to define the best chemotherapy regimens, but cisplatin- or carboplatin-based regimens are commonly used.
The patient was in partial response during the 12 months after chemotherapy and has made regular follow-ups to the outpatient department for 16 months.
In conclusion, we report on a 65-year-old woman with primary small cell carcinoma of the liver, who had a partial response to systemic chemotherapy.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print