임상시험 등록시스템 및 연구결과 보고지침
Registration and Reporting Guidelines for Clinical Trials
Article information
Trans Abstract
Selective outcome reporting is a major problem because it has a negative impact on our scientific knowledge and is unethical as it involves research on human subjects. And inadequate quality of trials may distort the results from systematic reviews and meta-analyses. Thus, Clinical medicine tries to solve the problem of trials by making public registration before patient enrollment mandatory and encouraging to report the research outcomes by a specific guidelines. In the past few years, the registration of clinical trials and the reporting system has become routine, supported by the International Committee of Medical Journal Editors. In Korea, the clinical research registry named ‘Clinical Research Information Service (CRiS)’ was recently established and became a data provider as a primary registry to the World Health Organization (WHO) International Clinical Trial Registry Platform search portal. To expand the registration system and to operate them successfully and comprehensively, active participations of researchers as well as the government supports are required. (Korean J Med 2012;83:309-318)
서 론
근래 임상 진료나 의료 정책과 같은 중요 의학적 행위를 수행함에 있어서 근거 기반의 결정이 강조되고 있고, 따라서 근거의 바탕이 되는 임상 연구의 중요성이 점점 더 커지고 있다. 잘못 수행된 연구에 의해서 엄청난 비용 낭비와 의료과오로 이끄는 정책이 결정될 수도 있지만, 잘 수행된 임상시험은 현재까지의 의학적 개념을 바꿀 수 있을뿐 아니라 특정 약제의 퇴출과 같은 제약 기업의 운명을 결정하기도 하는 강력한 힘을 발휘할 수 있다. 따라서 임상시험의 연구과정 및 결과 보고에 있어서 투명성, 윤리성과 객관성이 강조되는 것은 당연하며 이를 위한 노력으로 임상시험 등록과 보고지침을 개발 및 강화하는 국제적 노력들이 계속되어 왔다.
최근에 국제의학저널편집자위원회(International Committee of Medical Journal Editors, ICMJE)에서는 ‘생의학학술지에 투고하는 원고의 통일양식(Uniform Requirements for Manuscripts Submitted to Biomedical Journals)’을 통하여 모든 임상시험을 공공 차원에서 연구계획 단계에서부터 등록관리하는 것을 논문게제의 필수 요건으로 제시하고 있으며, 사전 등록되지 않은 임상시험에 대해서는 논문 게재를 거부하고 있다[1,2]. 또한 연구 방법 및 결과의 투명한 공개를 위해 기본적으로 갖추어야 할 요건을 제시한 여러 보고지침들을 개발하여 국제적 표준으로 지정하고, 선택한 연구방식에 따라 특정한 보고지침을 따르도록 하고 있다[2].
본 고에서는 현재 사용되고 있는 웹기반의 국제 및 국내 임상시험 등록시스템들을 소개하고, 보고 지침의 종류 및 개발 현황과 가장 흔히 사용되는 CONSORT (Consolidated Standards of Reporting Trials)와 STROBE (STrengthening the Reporting of OBservational studies in Epidemiology)에 대해 살펴보고자한다.
본 론
임상시험 등록시스템
임상시험 등록시스템의 필요성
약물이나 기기의 효과를 조사하고자 하는 임상연구를 수행하는 과정에서 연구의 규모가 크거나 후원을 받았거나 특히 특정 영리단체에서 연구가 시행되는 경우에 연구 결과가 의미가 있으면 보고하지만 결과가 나쁘게 나오면 보고하지 않는 유혹을 받는 경우가 발생한다. 또한 의미 있는 결과를 도출하기 위해 여러 가지 측정 도구 혹은 방법 중 결과가 의미 있게 나왔던 방법만을 선택하는 경우도 있다. 연구자 뿐만 아니라, 의학학술지 편집인들도 연구의 대상자 수가 적거나 결과가 유의하게 나오지 않는 경우 출판을 꺼리는 경우가 많다. 이러한 행위들을 선택적 보고(selective reporting)라고 하고[3], 가설에 합당하지 않는 연구 결과를 출판하지 않거나 일부 내용만 보고하게 되어 발생하는 삐뚤림을 출판 삐뚤림(publication bias)이라고 한다[4]. 이러한 잘못된 보고 행위는 광범위하게 시행되고 있었는데, 한 연구에 의하면 102개 임상 시험을 조사하였을 때 효과 관련 결과의 50%, 유해 관련 결과의 65%는 부적절하게 보고되었으며, 보고된 결과가 그렇지 않은 결과에 비해서 통계적으로 의미 있을 가능성이 월등하게 높았다[3,5]. 선택적 보고의 삐뚤림이나 출판 삐뚤림의 가장 큰 문제는 통계적 혹은 임상적으로 의미 없는 임상 결과들이 보고되지 않는 경우가 많기 때문에 이를 충분히 고려하지 않는다면, 임상 지침의 근거가 되는 메타분석 혹은 체계적 문헌고찰의 결과가 과대평가될 가능성이 크다는 것이다[6]. 따라서, 임상 연구가 시행되기 전부터 방법, 결과 변수 등이 공공적 차원에서 등록관리 된다면 삐뚤림을 방지하고 신뢰성 있는 결과들을 얻음으로써 중대한 의학적 의사결정에 도움이 될 것이라는 생각에서 임상시험 등록시스템이 제안되었다.
뿐만 아니라 임상시험 등록은 연구윤리적인 측면에서도 필요한데, 임상시험에 대한 정보 공개를 통해 연구자가 유의한 결과만 발표할 수 없도록 방지하고 불필요한 임상시험의 중복을 피할 수 있도록 하여 피험자의 권익을 보장하고, 연구 지원을 효율적으로 배분하도록 한다. 또한, 관련 연구자 및 기관에 세계적인 연구 동향 및 지식을 제공하고, 환자 및 일반인들에게는 편견없는 정보를 제공하여 임상시험의 참여 기회를 주게 되므로 오히려 대상 모집에 이득이 될 수도 있다[7]. 아직 상당수의 국내 연구자들이 연구정보의 보호 차원에서 임상시험등록을 꺼리고 있으나 임상연구의 윤리적 수행 및 투명성이 더욱 중요하다는 것이 현재의 국제적인 통념이다.
임상시험 등록시스템의 개발
임상시험 연구는 인간을 대상으로 한 연구인 만큼 그 결과가 질병관리 및 건강에 기여할 수 있어야 하므로, 기대하였던 바와 다른 결과가 도출된 경우에도 연구자는 솔직하게 그 결과를 공개할 의무가 있다. 이러한 문제점들을 해결하는 방법으로 제시된 것이 임상시험 등록(clinical trial registry)이다.
미국에서는 1997년 신약 개발을 위한 임상시험 정보등록을 법제화하는 과정에서 미국 국립보건원(National Institutes of Health, NIH)과 식품의약품안전청(Food and Drug Administration, FDA)의 공조로 임상시험 등록사이트인 Clinical Trial. gov가 개설되었고 2002년부터 임상시험등록이 본격화되기 시작하였다[8]. 이후 ICMJE에서는 2005년 7월부터 소속 회원 의학학술지(member journal)에 임상시험 논문을 발표하고자 한다면 피험자가 연구에 등록되기 전에 공적 기관에서 운영하는 등록시스템에 등록하는 것을 의무화하였으며, 사전 등록되지 않은 임상시험에 대해서는 논문 게재를 거부하도록 하였다[2]. 또한, 세계의사협회(World Medical Association, WMA)도 2008년 헬싱키선언 제6차 개정안에서 임상시험을 시작하기 전에 사전 등록할 것을 명시하였다[9]. 세계보건기구(WHO)에서도 연구의 투명성과 타당성을 강화하고, 과학적 근거의 수준을 높이기 위해 2005년 8월에 기준에 충족하는 각 나라 혹은 지역에서 운영되는 임상시험 데이터베이스를 primary registry로 선정하여 이들간 연계로 등록된 모든 임상연구를 통합 검색할 수 있는 국제임상시험등록 플랫폼(International Clinical Trial Registry Platform, ICTRP)을 구축하였다. 임상시험에 대한 국가간의 차별을 감소시키고 각국에서 일정 수준 이상의 임상연구 등록시스템 구축을 독려하며 등록 의무화 정책을 강화하도록 지원하기 위함이다. 임상시험의 사전등록의 의무화가 확산되고 있으며, 이제 사람을 대상으로 하는 모든 임상연구의 등록은 거스를 수 없는 흐름이 되었다[10].
현재 2012년 7월말 ClinicalTrials.gov.에는 각종 연구비 지원을 받는 미국 내의 약 130,000개의 임상연구 및 179개국의 연구가 등록되어 있고 하루 약 65,000명의 연구자와 일반인이 웹사이트를 방문하고 있다[8].
ICMJE가 요구한 등록시스템의 요건으로는 첫째, 별도의 비용 지불 없이 누구나 등록과 검색이 가능하며 둘째, 비영리 단체 또는 정부기관에 의해 운영되고 셋째, 등록 자료에 대한 검증과정이 체계화되어 있고 객관성을 유지하여야 한다. 임상연구 등록은 웹기반으로 이루어지는데, 현재까지 ICMJE가 인정하는 임상시험등록 기구로는 미국 NIH의 등록기구인 Clinicaltrials.gov를 비롯하여 ANZCTR (www.anzctr.org.au), ISRCTN register (www.ISRCTN.org), UMIN clinical trial registry (www.umin.ac.jp), Netherlands trial register (www.trialregister.nl)이며 2007년부터 WHO에서 운영하는 ICTRP (www.who.int/trialsearch)의 총 14개국의 primary registry들도 임상시험등록 기구들도 인정하고 있다[2]. WHO에서는 국가당 하나의 primary registry만 인정하고 있는데, 각 나라의 등록된 자료는 국제사회와 공유하게 된다. 2011년 6월 이후에는 EU clinical trial registry인 EudraCT (https://eudract.ema.europa.eu/)가 ICMJE의 인정 등록시스템으로 새롭게 추가되었다[10].
임상시험 등록시스템마다 요구하는 등록항목들은 유사하다. 우선 등록 이전에 각 기관의 임상시험심사위원회(Institutional Review Board, IRB)의 심의를 통과하여야 한다. 그 외 등록해야 하는 항목에는 1) 연구고유번호, 2) 중재나 비교대상에 대한 기술, 3) 연구가설, 4) 성과 측정방법 (primary and secondary outcome), 5) 연구대상자 선정기준, 6) 연구시작, 연구종료, 자료입력완료, 자료 분석 완료 예정일 등 주요일정, 7) 연구대상자 수, 8) 연구비지원 기관, 기업 또는 단체, 9)연구책임자에 대한 연락처 등의 정보가 포함되어야 있어야 한다(Table 1) [7].

임상시험 등록 요구의 확대 및 절차의 강화
ICMJE가 2007년에 채택한 WHO의 정의에 의하면, 임상시험(clinical trials)이란 사람을 대상으로 한 가지 혹은 그 이상의 중재 혹은 치료(예, 약물, 수술 기구, 행동 치료, 식이중재, 시술법)를 시행하여 그 건강 관련 결과를 평가하기 위한 전향적인 연구를 의미한다. 연구자가 중재를 결정할 수 없는 순수 관찰의 연구는 이 WHO의 정의에 의하면 임상시험에 속하지 않는다. 현재까지 무작위 대조군 시험(randomized controlled trial, RCT)의 중재연구에 비해 관찰연구에 대해서는 연구등록의 필요성이 덜 강조되었고, 논문 게재의 필수 요건으로 제시한 의학학술지도 많지 않았다. 그러나 많은 소속 학술지들이 2008년 이후 임상시험의 정의를 점차 확대하고 있기 때문에 그 기준이 모호한 경우 아예 등록할 것을 ICMJE에서도 권하고 있는 실정이므로, 사람을 대상으로 하는 연구라면 미리 등록시스템에 등록하는 것이 안전할 것이라 사료된다[2]. 이미 ClinicalTrials.gov를 비롯한 많은 등록 시스템에서도 사람을 대상으로 하는 모든 연구의 질 향상과 투명성을 지향하고 불필요한 연구의 중복을 막기 위해 관찰 연구 등록 또한 적극 권하고 있는 실정이며 점차 강조될 전망이다.
2007년에 발표된 미국 FDA의 시행령(FDAAA 혹은 US public law 110-85)에 의하면 자국 내에서 진행되는 약물 및 의료기구 관련 2-4상 임상시험에 대해서 연구정보뿐 아니라 부작용 및 1년 이내의 기초적인 결과까지 보고하도록 규정하고 있고 이를 어길 경우 연구비 중단 혹은 벌금 등의 강력한 규제를 명시하고 있다[11]. 아직까지 그 외 다른 나라 등록시스템에서는 연구결과에 대해 의무화하고 있지 않으나 차후 논문화 여부에 상관없이 시험 결과의 투명한 공개를 요구할 가능성은 점차 증가할 것으로 예상된다[7].
현재 국내학술지에서는 임상시험의 사전등록을 논문게재의 필수조건으로 규정하고 있지는 않으나, 대한의학학술지 편집인협의회를 중심으로 학술지에서 사전등록의 조건을 제시할 것을 권장하고 있으며, 국제적인 학술지로 도약하기 위해서도 이러한 요건의 도입은 필요할 것이다.
국내 임상시험의 등록: CRiS
최근까지는 영향력 있는 임상시험을 수행하려면 해외에서 운영하는 웹기반 임상시험 등록시스템에 등록을 해야 했는데 방법이 용이하지 않아, 이런 이유로 국내에서는 임상시험 등록의 관행이 정착되지 못했었다. 그 동안 질병관리본부 국립보건연구원에서는 임상연구정보서비스(Clinical Research Information Service, CRiS) 시스템을 구축하고 WHO ICTRP와 연계하는 작업을 추진하여, 2010년 5월에 드디어 WHO ICTRP의 11번째 primary registry로 가입하게 되었다[7]. 따라서 CRiS에 등록되는 모든 임상연구들이 WHO platform에 등재되므로 다른 국제 등록 사이트에 별도 등록없이 국제적 학술잡지의 인정을 받을 수 있게 되었다. 아직까지 국내에서는 모든 임상시험의 등록이 제도화되지는 않았으나, 식품의 약품안전청 및 보건복지부 연구개발사업에서는 등록을 권고하고 있다.
CRiS는 한 가지의 연구에 대하여 WHO ICTRP에서 요구하는 최소 필수항목을 포함하여 약 50개 항목을 등록받으며, 이 항목들은 총 11개영역(연구개요, 임상연구윤리심의여부, 연구자, 연구비 지원기관 및 책임기관, 연구현황, 연구요약, 연구설계 및 중재, 대상자선정기준, 결과변수 등)에 구성되어 속해 있다. 다른 임상시험 등록 사이트와 마찬가지로 임상 연구심의위원회 혹은 기관생명윤리위원회의 승인을 받은 과제에 한하여 등록이 가능하고, 첫 피험자 모집 전에 등록을 마쳐야 한다[12]. 국내 및 WHO ICTRP와 데이터 연계를 통한 국제적 정보 공개가 함께 이루어져야 하므로 국문과 영문으로 이중등록을 해야 하지만 실제 절차가 까다로운 편은 아니며 대부분 수일 내에 등록번호를 부여 받게 된다. 이미 다른 해외 등록기구에 등록이 되어 있더라도 CRiS 등록시 이를 표시하면 WHO ICTRP로 알리게 되어 이후의 자료를 CRiS에서 편리하게 관리할 수 있다. CRiS 역시 웹을 기반으로 하므로 시간과 공간에 제약없이 등록과 검색이 가능하며, 등록된 정보는 국내외 연구자는 물론 임상시험에 관심 있는 일반인에게도 공개된다.
2012년 6월말 현재 464여건에 달하는 임상연구가 등록되어있고, 이 중 중재연구가 325건, 관찰연구가 139건이다. 2010년 5월에 개설되고 2011년 8월에 등록 연구가 180건이 안되었던 점을 고려해보면 그 등록 증가 속도는 급격히 빨라지고 있다고 할 수 있다.
의학 연구 보고 지침
여러 분야의 연구자 및 편집인들이 모여 연구 과정 및 결과 보고의 투명성 등을 갖추기 위해 논문이 기본적으로 갖추어야 할 요건을 제시하였다. 무작위 대조군 연구의 보고를 위한 CONSORT (consolidated standards of reporting trials) 양식이 발표된 이후 이에 따라 작성된 보고의 내용과 질이 전보다 향상되자, 메타분석(meta-analysis)을 위한 QUOROM (quality of reporting of meta-analyses)와 MOOSE (metaanalysis of observational studies in epidemiology), 진단 정확도 연구(diagnostic accuracy studies)를 위한 STARD (standards for the reporting of diagnostic accuracy studies), 관찰연구의 보고 지침인 STROBE (strengthening the reporting of observational studies in epidemiology)가 개발되었다[13]. 최근에는 개발된 보고지침들을 더욱 세분화하고 확장한 지침들이 개발되었는데, STROBE에서 세분화된 유전자 관련 연구에 대한 STREGA (Strengthening the Reporting of Genetic Association Studies)와 QUOROM으로 부터 발전시켜 대체된 체계적 문헌고찰의 보고지침인 PRISMA (Preferred Reporting Items for Systematic Reviews)가 개발되었다[14,15].
2006년에는 다양한 보고지침들의 확대를 돕고 보고의 질 향상을 모니터 및 분석하여 지침을 수정 보완하는 등의 작업을 위해 Equator (Enhancing the Quality and Transparency Of health Research) network가 설립되었다[15]. 이 기구의 웹사이트를 통하여 여러 보고지침에 대한 자료를 좀 더 손쉽게 얻을 수 있고 최근의 개발 상황, 현 보고지침 관련 연구들을 부분적으로나마 검색할 수 있다. 또한 결과 보고를 준비하는 연구자들에게 보고지침 선택에 도움을 주는 내용들도 게시하고 있다.
국제 표준의 보고지침을 따르는 의학학술지의 범위는 점차 증가하고 있다. 이미 NEJM, Lancet, JAMA, BMJ와 같은 의학관련 핵심 학술지들은 논문 제출시 대부분 CONSORT, STARD, STROBE, PRISMA, MOOSE의 지침을 따를 것을 요구하고 있다(Table 2). 본 고에서는 이 중 흔히 사용되는 두 가지 보고지침인 CONSORT와 STROBE에 대해 살펴보고자한다.

무작위 대조군 연구의 보고를 위한 권고안-Consort statement
연구의 강점과 한계를 평가하기 위해서는 연구의 디자인과 방법의 절차가 투명하게 공개되어야 한다. 특히 무작위 배당 과정이 불완전하게 기술된 경우 논문의 결과를 객관화하여 적용하기에는 어려움이 따른다. 1990년대 이전에 보고된 무작위 대조군 임상시험은 기본 자료조차도 충분하게 제시되지 않은 연구가 많았다[16]. 한 가지 예로, 우울증에 대한 일차 선택 약물로서의 SSRI (selective serotonin reuptake inhibitor) 효과를 검증하기 위해 1990년대 후반까지의 무작위 대조군연구 122개의 논문을 분석한 연구에 의하면 이중 단 한 개의 논문만이 연구절차를 적절하게 기술하여 합의(consensus)를 도출하기에 불가능하였다[17].
이에 임상시험 보고의 질을 향상시키려는 임상연구자, 통계학자, 방법론 전문가, 의학 학술지 편집자 등으로 이루어진 독자적인 그룹의 논의가 있었고, 임상시험 보고를 위한 일정한 기준으로 32개의 체크리스트 항목을 정하고, 연구흐름도를 만들 것을 제안하였다(SORT statement). 여기에 다른 그룹에서의 권고안을 합쳐서 1996년에 도출하게 된 것이 CONSORT (CONsolidated Standards of Reporting Trials)이다 [18]. CONSORT 양식은 점차로 많은 의학 및 보건 분야 학술지와 국제의학저널편집자위원회, CSE (Council of Science Editors), WAME (World Association of Medical Editors)와 같은 편집인 협의회의 지지를 얻게 되었고, 2001년 개정안을 거쳐 CONSORT 2010 최신 개정판이 다시 보고되었으며 [19-21], 현재 임상시험 연구 보고의 통합된 표준으로 인정받고 있다. 현재 PubMed에 등록된 의학잡지의 50% 이상이 CONSORT를 지지하고 이에 따라 보고할 것을 제시하고 있다.
CONSORT 2010 양식에는 체크리스트 항목과 흐름도로 구성되어 있는데, 이는 일차적으로 임상시험 연구에 대한 논문 작성, 심사 및 평가에 사용하기 위한 것이다. 체크리스트에는 25개의 항목이 있는데, 연구제목, 서론, 연구방법, 결과, 고찰 등으로 나누어서 각각에서 제시해야 할 항목을 기술하고 있다(Table 3) [20]. 결과에서는 피험자에 대한 정보를 흐름도(participant flow)로 제시할 것을 권장하고 있는데, 이 흐름도에는 참가자 선정, 치료군 배정, 추적관찰, 최종 분석 대상의 내용이 포함된다(Fig. 1) [20,21]. 체크리스트를 수정하여 사용하는 것은 만들어진 과정이 다르기 때문에 추천되지 않고, Annals of Internal medicine, Lancet, BMJ 등의 CONSORT를 채택한 학술지 모두 양식을 그대로 이용하고 있다. 대부분 무작위대조시험에 적용할 수 있지만, 저자, 편집자, 독자들이 어떤 유형의 연구설계에 CONSORT statement를 적용 가능한지 궁금하다면 CONSORT 웹사이트에 질의할 수도 있다.

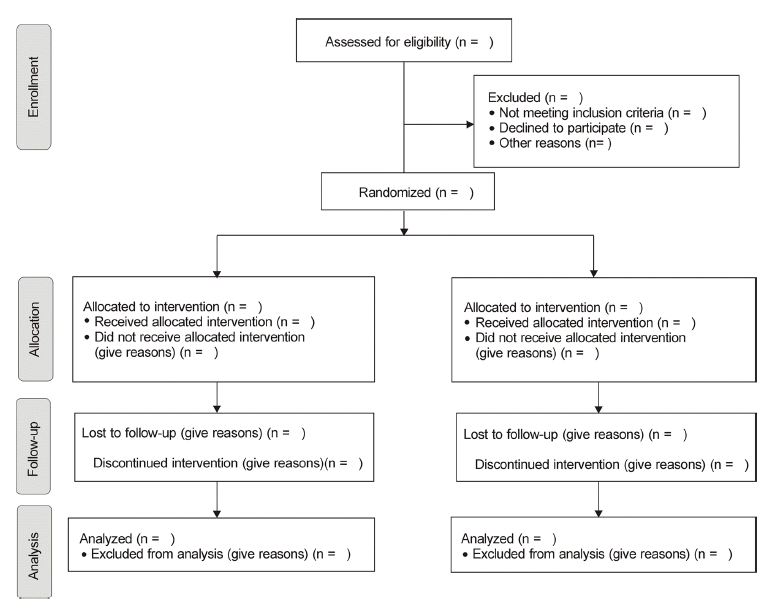
CONSORT을 사용한 십 년 남짓의 기간 동안 임상시험 결과의 부적절한 보고를 상당부분 줄일 수 있을 뿐 만 아니라, 연구의 진행 과정에도 긍정적인 영향을 주었다고 평가되었다[22,23]. 국제 연구비 지원기관 또한 이러한 영향을 인정하여 연구비를 신청할 때 계획서에 CONSORT 항목을 반영하도록 권장하고 있다. 그러나 주의할 점은 CONSORT는 무작위 대조군 연구의 방법과 결과를 세심히 기술하도록 함으로써 논문의 완성도와 투명성, 명확성을 높이지만, 논문이 전문 분야 내에서 얼마나 질적 가치가 높은지 혹은 흥미를 유발할만한 논리적인 흐름을 가지는지에 대한 평가와는 관련성이 없다는 것이다[13-20].
관찰 연구의 보고를 위한 권고안-STROBE statement
임상의학 연구에서 잘 고안된 무작위대조시험이 가치있음은 널리 알려져 있으나 모든 의학적 질문에 대하여 진행되기는 어려운 현실이다. 임상의학의 많은 연구들은 무작위 대조시험연구보다는 관찰연구의 형태로 보고되고 있고, 매우 유병률이 낮은 연구 등 연구 주제에 따라서는 관찰 연구가 더 적합한 것으로 인정받기도 한다. STROBE는 코호트연구, 환자-대조군 연구, 단면 연구와 같은 관찰연구 보고의 질을 향상시키고자 개발되었다. 여러 논문 분석에서 관찰연구의 보고도 무작위 배정 임상시험에서와 같이 대부분 정보가 적절히 제공되지 않았음에도 불구하고, 보고 양식의 체계화는 매우 늦어져 2004년 처음 개발되었다. 현재 STROBE는 2007년 10월에 개정된 4판이 쓰이고 있으며 국제의학저널편집자위원회를 포함한 100여개 이상의 학술지에서 이 권고안을 따르도록 저자들에게 요구하고 있다[13,14].
STROBE statement에는 코호트연구, 환자-대조군 연구, 단면연구에 따라, 각 연구마다 22개의 체크리스트 항목이 제시되어 있다[14]. 이중 18개는 연구형태와 상관없이 공통항목이고 나머지 4개의 항목은 연구형태에 따라 항목별로 차이가 있다. 일부 항목은 환자-대조군 연구에서 환자군과 대조군에 각각 따로 적용되고, 코호트연구에서는 각각 노출군과 대조군에 따로 적용되는 항목이 포함되어 있다. 연구제목, 서론, 방법, 결과, 고찰 내에서 다루어져야 할 항목들이 제시되어 있는데, 연구방법에서는 연구의 설계, 참여자, 변수, 자료 소스, 측정법, 분석방법 등이 포함되어 있고, 결과에서는 연구 대상자의 기본 특성, 성과지표(outcome) 등이 포함되어 있다. 사용에 관한 자세한 해설과 예시까지 담고 있어서 이용하기 편리하다. STROBE statement의 채택 후 역학적 관찰 연구의 질이 얼마만큼 향상되었는가에 대한 대규모 연구 보고는 아직 없지만, 연구의 투명성, 명확성을 바탕으로 하여 연구 결과를 더욱 비판적으로 해석할 수 있게 되었다는 보고가 있었다[24].
결 론
임상연구는 사람을 대상으로 하기 때문에 그 내용 및 과정에 있어서 엄격한 윤리성과 과학적 객관성이 요구된다. 연구 시작 전부터 임상시험등록을 하고, 절차를 투명하게 공개하며, 연구 결과를 표준화된 지침에 맞춰 보고하는 체계는 이제는 거스를 수 없는 흐름이 되었다. 힘겨운 작업이기 보다는 좀 더 양질의 연구를 수행하고, 검증된 연구 보고들를 보다 쉽게 선택할 수 있는 기회가 많아졌다는 생각이 더 옳을 것이다. 나아가 이러한 흐름은 의학적 진료지침(guideline) 마련 등의 중대한 의학적 의사결정에 큰 도움이 되리라 예상한다.
아울러, 국내 임상시험등록관리시스템의 구축으로 인해 국내 임상연구 수행의 정보를 국제적으로 공유함으로써, 국내 연구의 객관성 향상과 윤리적 수행뿐 아니라, 연구 수준의 우수함을 해외에 홍보하는 기회가 될 것으로 생각된다. 이러한 등록관리시스템의 성공적 운영을 위해서는 정부의 지원 뿐 아니라 민간차원, 특히 연구수행자들의 적극적 참여가 필요할 것이다.