소장 신경내분비암종을 동반한 Peutz-Jeghers 증후군 1예
A Case of Peutz-Jeghers Syndrome with Small Bowel Neuroendocrine Carcinoma
Article information
Abstract
PJS는 위장관의 다발성 과오종성 용종증과 피부 점막의 색소 침착을 특징으로 하며, 상염색체 우성으로 유전하는 드문 질환으로, 위장관 출혈이나 장폐색, 장중첩증 등의 합병증을 야기할 수 있다. PJS에서는 위장관 및 타 장기에서의 악성종양의 발생 빈도가 높으나, 위장관에서 보고된 종양의 경우 대부분 선암이며, PJS가 신경내분비종양과 동반된 경우는 아직 국내에 보고된 바 없다. 저자들은 PJS 환자에서 발생한 소장 기원의 소세포 신경내분비암종 1예를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
Trans Abstract
Peutz-Jeghers syndrome (PJS) is an autosomal dominant inherited disorder characterized by hamartomatous polyps in the gastrointestinal tract and mucocutaneous melanin pigmentation. Hamartomas are not generally regarded as premalignant, although patients with PJS are at increased risk for common and unusual types of gastrointestinal and non-gastrointestinal malignancies. However, most of the reported gastrointestinal malignancies have been adenocarcinomas, and few reports of an association of this syndrome with a neuroendocrine tumor (NET) have been published. Moreover, no case of this syndrome with NET has been reported in Korea. Here, we report a 21-year old male with PJS who had a small bowel neuroendocrine carcinoma. (Korean J Med 2013;84:698-703)
서 론
Peutz-Jeghers 증후군(Peutz-Jeghers syndrome, PJS)은 위장관의 과오종성 용종증과 피부 점막의 색소 침착을 보이는 유전 질환으로, 1921년 Peutz에 의해 처음 보고되었으며 1949년 Jeghers 등에 의해 상염색체 우성으로 유전되는 것으로 밝혀졌다. 발병률은 120,000명당 1명이며, 거의 모든 인종에서 발견되고 남녀의 발생 빈도 차이는 없다고 한다. 용종은 위장관의 어느 부위에서나 발생할 수 있으며, 소장 특히 공장에서의 발생률이 가장 높고 다음으로 대장, 위, 십이지장 순이며, 이 외에도 방광, 요도, 기관지 및 비강에서도 발생한다고 보고되었다. 합병증으로 용종의 크기 증가에 따른 장폐색과 장중첩증, 급, 만성 위장관 출혈 등이 흔히 발생할 수 있다. 과오종은 이 자체가 암의 전구단계로 여겨지지는 않으나 PJS에서는 비교적 젊은 나이에 위장관 및 타 장기에서의 악성종양의 발생 빈도가 높은 것으로 알려져 있으며, 한 연구에서는 정상인에 비해 종양 발생의 위험이 15배나 높은 것으로 보고하였다[1].
신경내분비종양(neuroendocrine tumor, NET)은 위장관, 기관지, 담관계, 난소, 고환, 흉선 등에 존재하는 신경내분비계 세포에서 유래하며, 74%가 위장관계에서 발생하고 25%는 호흡기계통에서 발생한다. 위장관별 발생 빈도는 소장, 충수, 직장, 대장, 위의 빈도로 발생한다[2]. 이 중 소세포 신경내분비암종(small cell neuroendocrine carcinoma)은 폐 이외의 장기에는 드물게 발생하여 전체 소세포암 중 4% 미만을 차지하며, 발견 당시 주변 장기침윤과 전이가 동반된 경우가 많아 예후가 매우 불량하다. 또한 신경내분비암종을 진단하는 데 있어서 면역 조직화학 검사가 유용하여 chromogranin A, synaptophysin, CD56, neuron-specific enolase 등에 양성 반응을 보이나 모든 신경내분비 표지자에 일관되게 염색되는 경우는 많지 않다.
PJS 환자에서 보고된 종양의 경우 대부분 위장관 선암이었으며[1,3], PJS가 신경내분비종양과 동반된 경우는 아직 국내에 보고된 바 없다. 이에 저자들은 PJS 환자에서 발생한 소장 기원의 소세포 신경내분비암종 1예를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
증 례
환 자: 남자, 21세
주 소: 좌하복부 통증과 흑색변
현병력: 6개월 전부터 발생한 간헐적인 복통과 2개월 전부터 발생한 흑색변이 심해져 내원하였다. 환자는 4개월 전 시행한 소장 조영술과 한 달 반 전 시행한 상부위장관 내시경, 대장내시경 검사에서 다발성 용종증 외에 특이 소견을 보이지 않았으며(Fig. 1), 한 달 전 시행한 99mTc-RBC scan 에서 출혈병소가 관찰되지 않아 경과 관찰 중이었다.
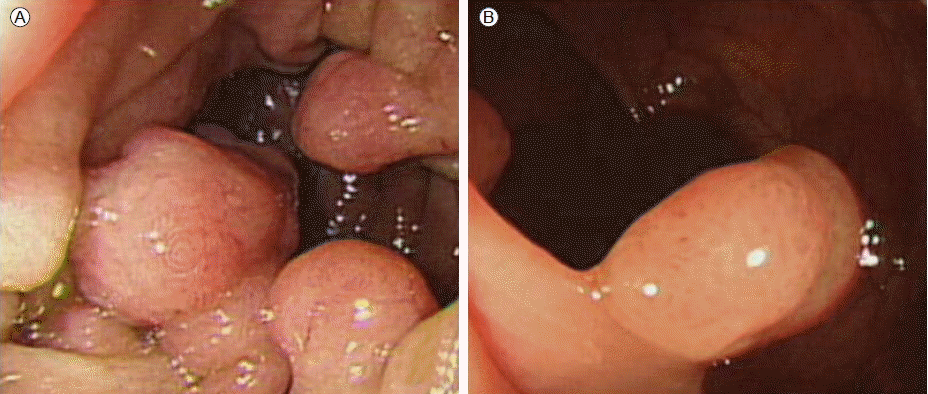
(A) Esophagogastroduodenoscopy showed multiple sessile or pedunculated polyps in the duodenum. (B) Colonoscopy showed multiple polyps in the sigmoid colon.
과거력: 생후 1년 후부터 구강 점막의 색소 침착이 있었으며, 10년 전 장중첩증으로 소장 부분 절제술을 시행받았다. 당시에 시행한 상부위장관 내시경, 소장 조영술, 대장내시경 검사에서 위장관에 다발성 용종증이 있었고 절제한 용종의 조직 검사 결과 과오종에 합당한 양상을 보여 PJS로 진단되었다. 그 외 만성 B형 간염으로 16개월 동안 adefovir를 복용 중이었다.
가족력: 형제는 없었으며, 부친이 입술 점막에 색소침착이 있었고, 9세 때 장중첩증으로 수술을 시행받은 적이 있었다.
사회력: 음주력과 흡연력은 없었다.
진찰 소견: 혈압 110/70 mmHg, 맥박수는 분당 104회, 호흡수는 분당 20회, 체온 38.5℃였으며, 안면은 창백하고 급성 병색을 보였다. 입술과 구강 점막, 손가락의 다발성 색소침착이 있었고(Fig. 2), 흉부 청진상 심음과 호흡음은 모두 정상이었다. 복부 진찰에서 좌하복부에 15 × 10 cm 크기의 딱딱한 종괴가 촉지되었고, 주위 조직에 고정되어 있었으며 압통과 국소적인 열감을 동반하고 있었다. 장음은 항진되어 있었다.
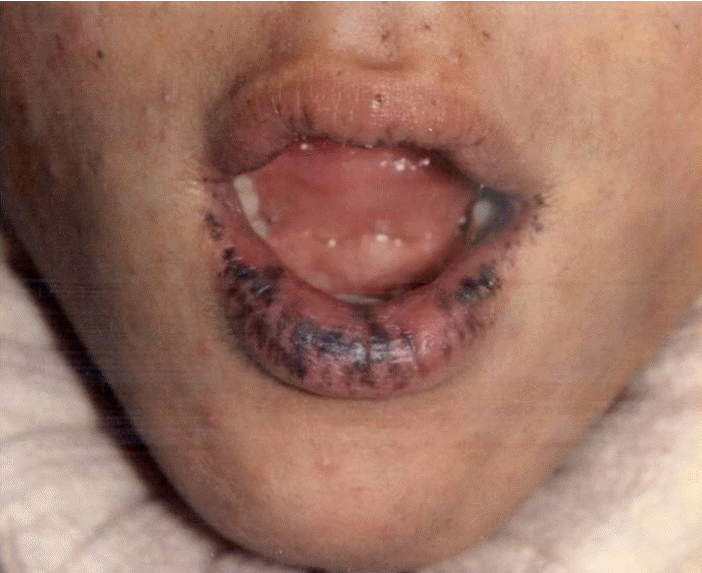
Mucocutaneous pigmentation of the lips.
검사실 소견: 말초혈액검사에서 백혈구 16,250/mm3, 혈색소 9.7 g/dL, 혈소판 349,000/dL, 혈청 생화학 검사에서 총 단백질 5.9 g/dL, 알부민 3.2 g/dL, AST/ALT 55/13 U/L, 총 빌리루빈 0.7 mg/dL, BUN 10 mg/dL, 크레아티닌 0.9 mg/dL, Na 132 mmol/L, K 4.2 mmol/L이었고, 면역혈청 검사에서 CRP 17.49 mg/dL였다. 암표지자 검사는 AFP 0.95 ng/mL, CEA 0.50 ng/mL 이하, CA 19-9 2.00 U/mL 이하로 정상 소견이었다.
영상의학 소견: 복부 전산화 단층촬영에서 불균일한 조영증강을 보이며 중심부 괴사를 동반한 15 cm 크기의 종괴가 좌하복강에 관찰되었으며, 소장과 장간막을 침범하고 있었고, 간전이 및 복막전이 소견을 보였다. 종괴의 크기가 매우 커서 원발 병소를 정확히 알기는 어려우나, 공장 내강의 동맥류성 확장(aneurysmal dilatation)과 전층이 비후된 소견으로 미루어, 소장에서 기원한 악성종양의 가능성이 높았다(Fig. 3).

Abdominal computed tomography showed (A) a 15-cm heterogeneous enhancing mass with central necrosis in the left abdominal cavity. (B) A 7-cm well-demarcated inhomogeneous mass in the right lobe of the liver, suggesting hepatic metastasis.
수술 소견: 향후 기계적 폐쇄 가능성이 높아 외과에서 개복 수술을 시행하였고, 개복 시 15 × 12 cm 크기의 소장 기원의 종괴가 관찰되었으며, 딱딱하고 출혈 경향이 있었다. 이 종괴는 하행결장을 침범하고 있었고, 장간막과 후복막에 단단하게 고정되어 Treitz 인대를 확인할 수 없었으며 복막전이 소견을 보였다. 부대동맥 림프절, 대동정맥 림프절, 장간막 림프절, 상장간막동맥 기시부 주변의 림프절 종대가 관찰되었다. 암의 진행이 심하여 근치적 수술이 불가능하다고 판단되어 장간막 림프절에 대한 조직 검사와 위-공장 문합술 및 영구 회장루를 시행하였다.
병리 검사 소견: 림프절에 침윤한 종양 세포는 작고 둥근 형태로 과염색상 및 10개의 고배율 시야에서 24개의 높은 유사분열 수를 보였고, 면역염색에서 chromogranin A, synaptophysin 양성 소견을 보여 신경내분비계 악성종양이 의심되었다. 이후 환자는 타병원으로 전원하여 간의 전이암 병소에 대해 경피적 간생검을 하였으며, 장간막 림프절에 침윤한 종양 세포와 동일한 소견을 보였고, 면역염색에서 pancytokeratin 양성 소견을 보여 소세포 신경내분비암종으로 진단되었다(Fig. 4).
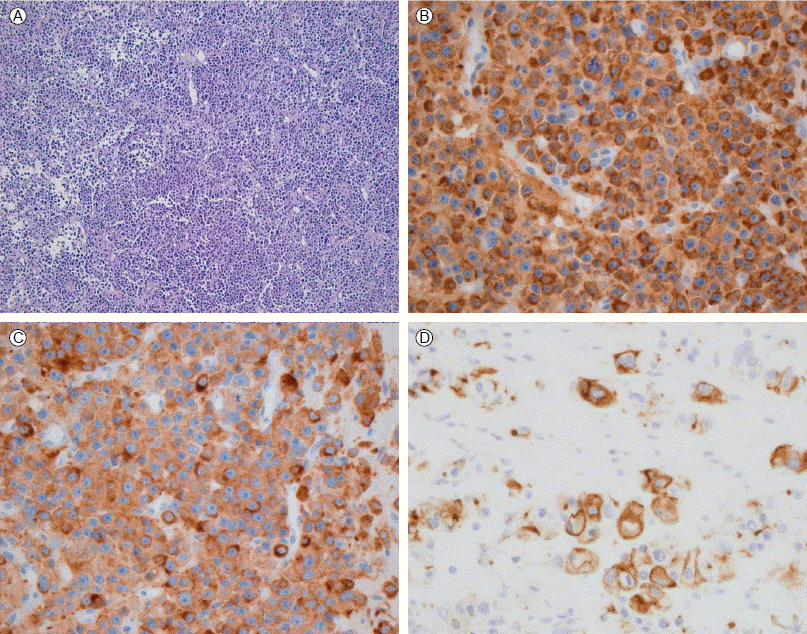
Histologically, (A) the mesenteric mass is composed of uniform, small round neoplastic cells with hyperchromatic nuclei and frequent mitotic figures (H&E, × 100). The neoplastic cells of the mesenteric mass are immunoreactive for chromogranin A (B) and synaptophysin (C) (immunohistochemical stain, × 400). (D) The tumor cells in the liver are immunoreactive for pancytokeratin (immunohistochemical stain, × 400).
치료 및 경과: 환자는 항암 약물 치료를 시행하였으나, 3개월 후 호중구 감소증과 동반된 폐렴으로 사망하였다.
고 찰
PJS에서 동반되는 종양으로 소화기관의 선암 발생이 빈번하며, 대장과 위를 비롯하여 십이지장, 공장, 회장에서의 발생이 보고되었다[4]. 하지만 이러한 소화관 선암의 발생에 대해서는 과오종성 용종의 악성 변화인지 de novo에 의한 것인지 아직은 불명확한 실정이다. 1982년 Perzin 등[5]이 과오종성 용종에서 선종성 변화 및 암종성 변화를 처음으로 보고한 이래, 악성종양 발생과 밀접한 관련이 있는 것으로 밝혀지고 있으며, Foley 등[3]은 과오종성 용종을 전암성 병변으로 간주하고 증상이 없는 환자에서도 용종 절제술을 권고하였다. 이 외에도 PJS에서 췌담관 악성종양, 양측성 유방암, 난소 및 자궁 경부암, 고환암, 폐암, 흑색종, 갑상선암, 비강의 편평세포암 등이 동반될 수 있다. 144명의 PJS 환자를 추적관찰하였던 연구에서는 32%에서 악성종양이 발생하여 일반인에 비해 암 발생률이 9배, 사망률은 3.5배 높다고 보고하였다[4]. 다른 연구에서는 15세에서 64세 군에서 악성종양 발생의 상대적인 위험도는 15.2배이며, 소장, 위장, 췌장, 대장 순으로 장기별 위험도가 높다고 보고하였다[1].
신경내분비종양이 동반되는 질환으로는 제1형 다발성내분비선종증(multiple endocrine neoplasia, MEN), 제1형 신경섬유종증(neurofibromatosis), 다발성 경화증(tuberous sclerosis), von Hippel Lindau 병 등이 알려져 있다. 이 질환들은 상염색체 우성으로 유전되고, 종양 억제 유전자의 결함과 연관되어 발생한다는 점에서 PJS와 유사성을 가진다. 제1형 다발성내 분비선종증은 기관지, 흉선, 위 등에서 신경내분비종양을 동반할 수 있으며, 폐 대세포신경내분비암종을 동반한 증례가 국내에서도 보고된 바 있다[6]. 또한 제1형 신경섬유종증 환자에서는 신경내분비종양의 유병률이 일반 인구에 비해 높은 것으로 알려져 있으며, 십이지장의 파터팽대(ampulla of Vater)에서 흔하게 발생한다고 보고되었다. 그 외 크론병, 궤양성대장염과 같은 염증성 장질환과 다운 증후군 등에서 신경내분비암종의 동반 발생이 보고된 바 있다.
이번 증례에서 환자는 만성 B형 간염으로 adefovir를 복용중이었다. 만성 B형 간염 환자에서 간의 원발성 신경내분비 종양이 증례로 보고된 바 있으나[7], 현재까지 만성 간염 및 adefovir 복용과 신경내분비종양 사이에 연관성이 입증된 연구나 위장관에서 신경내분비종양의 동반 발생에 대한 증례는 없는 실정이다.
국내에서는 PJS에서 소화관 선암을 제외한 악성종양의 발생은 드물게 보고되고 있으며[8], 신경내분비종양이 병발된 경우는 아직 보고된 바가 없다. 국외 문헌을 보면, 1998년 Wada 등[9]이 16세 남성 PJS 환자에서 직장에 위치한 6 mm 크기의 점막하 종양에 대해 내시경 점막 절제술을 시행하여 직장 유암종(carcinoid)의 발생을 처음 보고하였다. 이후 최근 Chen 등[10]이 42세 남성 PJS 환자에서 십이지장에 발생한 9 cm 크기 저분화 신경내분비암종(poorly differentiated neuroendocrine carcinoma)에 의한 장중첩증을 증례로 보고하였으며, 환자는 수술적 절제를 시행하였으나 국소전이와 간전이가 발생하여 수술 5개월 후 사망하였다. 이상의 두 증례와 이번 증례를 비교해보면 PJS에서 신경내분비종양은 비교적 젊은 나이의 남자 환자에서 위장관에 발생했다는 공통점이 있으나, 이번 증례에서는 공장에서 기원한 15 cm 크기의 종양이 하행결장을 침범하고 있었고 개복 수술을 시행하였으나 암의 진행이 심하여 근치적 절제가 불가능하였다.
PJS에서 종양 발생은 종양 억제 유전자인 STK11의 결함과 연관된 것으로 알려져 있다. 이는 chromosome 19p13.3에 위치하며, 23Kb의 길이를 가진 9개의 exon으로 433개의 아미노산을 암호화한다. STK11은 악성화 작용뿐 아니라 혈관생성에도 관여하며 암세포주에서 과발현되었을 때 세포 주기 중 G1 시기를 정지시킴으로써 세포의 성장을 억제하는 것으로 알려져 있다. 따라서 STK11 유전자의 돌연변이 발생 시 암억제 역할에 제한을 받게 된다. 그러나 이번 증례에서는 STK11에 대한 유전자 검사는 시행하지 못하였다.
PJS에서 종양 발생에 대한 추적검사로 2년마다 소장조영술과 함께 상부위장관 내시경과 대장내시경을 시행할 것이 권고되며, 최근 비디오 캡슐 내시경이 소장의 종양 발견에 유용하다. 여성 PJS 환자에서는 유방암의 경우 25세부터 정기적인 검진을 시작하며 30세 이상에서는 매년 유방촬영 등의 유방 검사를 시행하고, 부인과적 영역의 악성종양에 대한 추적검사로 1년마다 골반초음파 및 자궁경부 도말법과 CA 125 검사를 권고하고 있다. 남성 PJS 환자에서는 사춘기 이전에 고환암의 발생률이 높으므로 이에 대한 정기적인 검사가 필요하다[4]. 그러나 이번 증례에서는 4개월 전 시행한 소장 조영술 검사에서 다발성 용종증 외에 특이 소견을 보이지 않았음에도 불구하고 내원 후 시행한 복부 전산화 단층촬영에서 15 cm 크기 소장 종양의 간전이, 복막 전이 소견을 진단하였다. 악성종양에서 흔히 동반되는 복통, 오심, 구토 등의 증상이 PJS 환자에서 용종이나 장중첩증에 의한 증상으로 오인되어 악성종양의 진단이 지연될 수 있으므로, 특히 증상이 있는 젊은 환자에서는 더 적극적인 검사를 시행해야 할 것이다. 또한 PJS로 진단받은 모든 환자에 있어 진단 당시나 향후 악성질환 발생의 가능성을 염두에 두고 소화관 및 비소화관에 대한 면밀하고 정기적인 추적검사를 시행하는 것이 필요하다.